Chemical Science | wSDTNBI - a novel network-based inference method for virtual screening

It is well known that Virtual Screening (VS) is a mainstream technique for rapid access to lead compounds, which is usually divided into two categories: Structure-based VS and Ligand-based VS. These methods have had many successes in lead compound discovery, but there are still some shortcomings. For example, structure-based VS methods rely on the 3D structure of the target, while ligand-based VS methods require predefined pharmacophore models of the ligand, etc. These shortcomings limit the scope of application of these virtual screening methods.
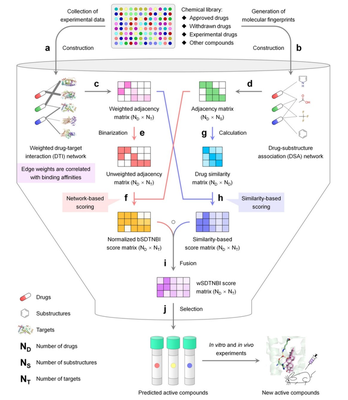
In recent years, with the rapid development of Systems Biology and Network Pharmacology, the intricate network relationship between drugs and targets has been gradually revealed, and the drug discovery model has gradually shifted from the linear model of “a drug → a target → a disease” to a network model of “multiple drugs → multiple targets → multiple diseases”. A series of network-based drug-target interaction prediction methods have been developed, namely Network-based Inference (NBI) and the subsequent substructure-drug-target NBI (SDTNBI and bSDTNBI). These methods can predict potential targets not only for known drugs and New Chemical Entity (NCE), but also for potential active compounds for targets of interest. Therefore, researchers tried to apply this network-based approach to virtual screening studies and proposed a new concept of Network-based VS (Network-based VS) with good results in preliminary case studies. These methods only require the construction of drug-target interaction networks based on known Ki, Kd, IC50, or EC50 values, without the need for known 3D structures of the targets or pharmacophore models of the ligands, and thus have a broader scope of application in a fast pace; and they allow virtual screening of multiple targets simultaneously, thus predicting the multidirectional pharmacological (polypharmacology) properties of potentially active compounds, which are not available in the aforementioned structure-based and ligand-based virtual screening.
However, existing network-based methods such as bSDTNBI can only predict the presence or absence of interactions between compounds and specific targets, and have not yet considered the strength of the interactions, thus making it difficult to effectively predict compounds with high activity against specific targets. To overcome this limitation, a new method called weighted substructure-drug-target network-based inference (wSDTNBI) was proposed. As a network-based method, wSDTNBI not only maintains the existing advantages of such methods, but also reasonably introduces binding affinity data that can reflect the strength of drug-target interactions, through a “two-pronged” approach that combines network inference and similarity inference. By combining network inference and similarity inference in a “two-pronged” approach, the predictive scoring can reflect the strength of the interaction and lead to rapid and efficient discovery of highly active compounds. The wSDTNBI method took only tens of seconds to complete the interaction prediction between more than 13,000 compounds and nearly 2,000 human targets, i.e., a virtual screening was conducted for these targets simultaneously. By conducting multiple types of in vitro experiments on some of the predicted results, seven new retinoic acid-related orphan receptor γt (RORγt) inverse agonists were successfully identified. Among them, two natural products, ursolic acid and oleanolic acid, were particularly active, with IC50s of 10 nM and 0.28 μM, respectively, against the predicted target RORγt. In this study, the binding constants of the active compounds to the target proteins were obtained by isothermal calorimetric titration experiments, and the structural features of the interaction between the active compounds and the target proteins were resolved by the crystal structure of the complexes. In addition, ursodiolic acid and oleanolic acid showed good therapeutic effects in animal experiments and are expected to be lead compounds for targeting RORγt in the treatment of autoimmune diseases such as multiple sclerosis.
Compared with recent structure-based or deep learning-based virtual screening studies for the same target, this study not only has a higher success rate, but also helps to obtain higher active potential drug molecules. Currently, the wSDTNBI method has been integrated into NetInfer (http://lmmd.ecust.edu.cn/netinfer), an online service system, for free use by users all over the world, which can help in drug discovery studies for multiple types of targets against different diseases.
Reference
- Wu Z, Ma H, Liu Z, Zheng L, Yu Z, Cao S, Fang W, Wu L, Li W, Liu G, Huang J, Tang Y. wSDTNBI: a novel network-based inference method for virtual screening. Chem. Sci., 2022, in press.
- Scior T, Bender A, Tresadern G, Medina-Franco JL, Martinez-Mayorga K, Langer T, Cuanalo-Contreras K, Agrafiotis DK. Recognizing pitfalls in virtual screening: a critical review. J. Chem. Inf. Model., 2012, 52(4): 867-881.
- Wu Z, Li W, Liu G, Tang Y. Network-based methods for prediction of drug-target interactions. Frontiers in Pharmacol., 2018, 9: 1134.
- Cheng F, Liu C, Jiang J, Lu W, Li W, Liu G, Zhou W, Huang J, Tang Y. Prediction of drug-target interactions and drug repositioning via network-based inference. PLoS Comput. Biol., 2012, 8(5): e1002503.
- Wu Z, Cheng F, Li J, Li W, Liu G, Tang Y. SDTNBI: an integrated network and chemoinformatics tool for systematic prediction of drug-target interactions and drug repositioning. Briefings in Bioinformatics, 2017, 18(2): 333-347.
- Wu Z, Lu W, Wu D, Luo A, Bian H, Li J, Li W, Liu G, Huang J, Cheng F, Tang Y. In silico prediction of chemical mechanism-of-action via an improved network-based inference method. British J. Pharmacol. 2016, 173(23): 3372-3385.
- Wu Z, Lu W, Yu W, Wang T, Li W, Liu G, Zhang H, Pang X, Huang J, Liu M, Cheng F, Tang Y. Quantitative and systems pharmacology. 2. In silico polypharmacology of G protein-coupled receptor ligands via network-based approaches. Pharmacol. Res., 2018, 129: 400-413.
- Wu Z, Wang Q, Yang H, Wang J, Li W, Liu G, Yang Y, Zhao Y, Tang Y. Discovery of natural products targeting NQO1 via an approach combining network-based inference and identification of privileged substructures. J. Chem. Inf. Model., 2021, 61(5): 2486-2498.
- Wu Z, Peng Y, Yu Z, Li W, Liu G, Tang Y. NetInfer: a web server for prediction of targets, therapeutic and adverse effects via network-based inference methods. J. Chem. Inf. Model., 2020, 60(8): 3687-3691.
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.