Nature | Synthon-based ligand discovery in virtual libraries of over 11 billion compounds

Virtual screening of potential hit compounds from molecular libraries is a common tool for early drug discovery, and the size and diversity of molecular libraries are critical to the hit rate of virtual screening. Currently, it is widely believed that there are more than 1020-1063 drug-like molecules in chemical space, and the size of the existing molecular libraries is only the tip of the iceberg, and the number of molecules and molecular backbone diversity still need to be improved urgently. One of the effective ways to expand the size of molecular libraries is to build virtual molecular libraries, and Enamine’s REAL (readily available for synthesis) is one of the most widely used virtual libraries today. This virtual library utilizes modular parallel synthesis (optimized reaction types) and molecular blocks, and has been scaled up to over 21 billion molecules. The virtual library is also fast (less than 4-6 weeks), reliable (>80% success rate), and relatively affordable. However, as the number of molecules in the virtual library skyrockets, the computational time and cost becomes a bottleneck for conventional docking-based virtual screening algorithms. For example, docking 10 billion compounds at 10 seconds/molecule would require more than 3000 years of running on a single CPU core, which could cost more than $800,000 if it goes to the cloud!
To keep up with the explosive growth in the number of virtual molecular libraries on the one hand, and to significantly reduce computational time and cost while maintaining docking accuracy and not losing the best scored compounds on the other, the industry is eagerly awaiting a new generation of fast screening methods for virtual molecular libraries. Recently, researchers have collaborated to propose V-SYNTHES (Virtual SYNThon Hierarchical Enumeration Screening), a modular synthon-based efficient screening method, significantly reduces the computational time and cost required for virtual screening without compromising docking accuracy. The key to the success of V-SYNTHES is to “reduce the whole molecule to zero” - instead of docking and scoring the complete molecule one by one, the molecule is broken down into a backbone and several synthons, and after finding a synthon that matches one part of the protein target pocket, then finding a synthon that matches another part of the target pocket. After finding a synth that matches one part of the target pocket, then finding another synth that matches another part of the target pocket, and so on, the complete molecule fits the bill. They validated the effectiveness of the V-SYNTHES method by targeting a cannabinoid receptor antagonist. By iteratively docking less than 2 million compounds, V-SYNTHES completes the screening of the 11 billion size Enamine REAL Space library more than 5000 times faster than conventional standard docking algorithms. After experimental validation, V-SYNTHES predicted novel cannabinoid antagonists with a hit rate of 33%, with 14 molecules reaching sub-muM levels of activity.
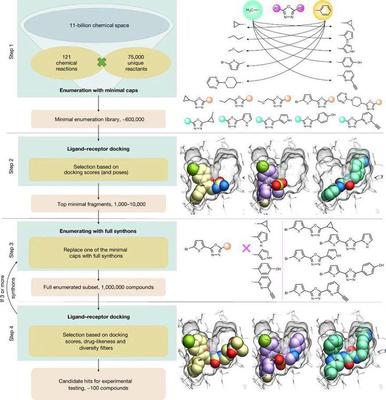
Workflow of V-SYNTHES
The workflow of V-SYNTHES is shown in the following diagram: (1) A fragment library, called the minimal enumeration library (MEL), is generated that can represent all possible backbone - synthons combinations in all reactions. Since only one of the synthons (i.e., the R group) is fully enumerated, the size of the MEL library is of the same order of magnitude as the synthons, with only about 600,000 MEL molecules. (2) Docking of MEL molecules to target receptors using flexible docking. A few thousand MEL molecules with the top ranking of docking scores were selected for the next enumeration step. Also, for more diverse results, the contribution of a particular reaction was restricted to no more than 20%. (3) Traverse another synton to obtain the complete compound, and this step produces a subset of compounds comprising roughly one million molecules. (4) Docking with the target receptor again, and screening based on docking fraction, drug-like properties, chemical diversity, etc. Of course, the above workflow example is for two synthons, and if the molecule contains more synthons, it is sufficient to repeat steps 3 and 4 until the complete compound is obtained. The top ranked compounds are further screened for PAINS, physicochemical properties, drug-like properties, novelty and chemical diversity, and finally 50-100 compounds are selected for synthesis and experimental testing.
Summary
In this paper, scientists introduced a new virtual molecular library screening method, V-SYNTHESIS, which can perform hierarchy-based screening of 11 billion virtual libraries and rapidly screen the highest scoring compounds by docking only a small fraction (<0.1%, ~2 million compounds) of the virtual molecular library, speeding up the screening of 11 billion virtual molecular libraries by more than 5000 times. Experimentally validated, V-SYNTHES predicts a 33% hit rate for novel cannabinoid antagonists (14 molecules reach the sub-μM level), doubling the success rate of a standard virtual screen based on a 115 million virtual molecular library (33% vs. 15%, and the latter is approximately 100 times more computationally expensive than V-SYNTHES), greatly improving the efficiency of the virtual screen. The potency, affinity (highest Ki reached 0.9 nM) and CB2/CB1 selectivity (50-200 fold) of the molecules were further improved by synthesizing analogues.
In addition, V-SYNTHES can also be used to discover potent antagonists of the kinase target ROCK1 (IC50 = 6.3 nM) with a hit rate of 28.5%. These data further support the application of V-SYNTHES for the discovery of lead compounds in different kinds of protein targets. The computational cost of the method increases only linearly with the number of synthons, is easily scalable to rapidly growing combinatorial libraries, and may be applicable to any docking algorithm.
Conceptually, V-SYNTHES utilizes a similar paradigm to FBDD, but the differences in details are still relatively obvious. (1) Classical FBDD requires testing the binding constants of fragments by highly sensitive methods (e.g. NMR or SPR), while V-SYNTHES undoubtedly relies heavily on the accuracy of docking scoring. (2) FBDD is generally limited to smaller molecular libraries (~1000 compounds) with smaller fragments (<200 Da), whereas V-SYNTHES uses relatively slightly larger (250-350 Da) and rigid initial MEL molecules, and most docking algorithms perform relatively well for such molecules. (3) The extension and ligation of fragments in FBDD often require relatively complex chemical reactions. In contrast, V-SYNTHES avoids the testing and complex synthesis of weakly bound fragments by performing fragment traversal in a very large but well-defined chemical space (optimized reactions), and the resulting dominant fragments are easy to optimize subsequently.
Reference
- Sadybekov, A.A., Sadybekov, A.V., Liu, Y. et al. Synthon-based ligand discovery in virtual libraries of over 11 billion compounds. Nature (2021).
- Now scientists can efficiently screen billions of chemical compounds to find effective new drug therapies.
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.