Science | Computed structures of core eukaryotic protein complexes

Proteins act as the scaffold and main substance constituting human tissues and organs and the protein-protein interaction (PPI) between protein monomers is crucial. The complex formed by the paired operation of proteins is an important foundation for the completion of various life activities. Therefore, the in-depth study of protein interaction is a necessary prerequisite for understanding and understanding various life phenomena. Among them, analyzing the structure of protein interactions is essential for understanding the function of protein interactions. However, there is still a lack of understanding of the three-dimensional structure of most protein complexes. The main obstacle is the instability of a large number of protein structures. The emergence of two AI algorithms, AlphaFold and RoseTTAFold, gave the hope of a breakthrough.
Both RoseTTAFold and AlphaFold can accurately predict protein structure based on the sequence, but their strategies are different. On the basis of the two-track neural network adopted by AlphaFold, RoseTTAFold adds a third track, which enables it to take into account different dimensions of information such as sequence, amino acid interaction, and three-dimensional protein structure at the same time. In addition, during the development process, RoseTTAFold’s team also tried a fast dual-orbit model. Although the accuracy of this dual-orbit model is not as good as that of the three-orbit model and AlphaFold, the calculation speed is nearly 100 times that of AlphaFold.
It is the different nature of the two sets of tools that give the research team the opportunity to combine the advantages of the two to screen for possible interacting proteins and predict their structure within the scope of the whole proteome. The dual-track RoseTTAFold is faster, which allows it to adapt to the scale of the whole proteome. In addition, RoseTTAFold focuses more on the co-evolutionary information in the protein sequence-proteins that can really interact in the cell should be co-evolved. AlphaFold is more focused on three-dimensional structure, so it is suitable for simulating whether two proteins can form a stable complex.
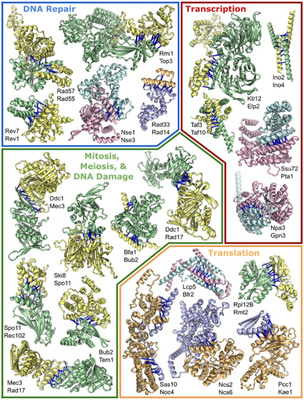
In this study, Prof. David Baker’s group extended the application scope of these AI tools for protein monomer structure prediction to predict the structure of protein complexes and predict those proteins that may interact in the cell within the entire proteome.
In this publication, they applied these methods to yeast, because yeast is an important eukaryotic model organism, and a large amount of experimental data on protein interactions in yeast can be used to measure the accuracy of their results. They used the co-evolution analysis of whole proteome amino acids and deep learning-based structural modeling to systematically identify and construct the accurate structure of yeast protein complexes. Their work uncovered 1,505 possible protein complexes. The three-dimensional structures of 699 of them have been analyzed by experimental methods, verifying the practicability of their methods. However, the three-dimensional structures of 700 predicted protein complexes are unknown, and 106 have never been described.
In order to discover protein function from these unresearched or unknown protein-protein interactions, scientists around the world are working together. Combined with the three-dimensional structure model generated in current research, these collaborations have deepened understanding of organisms in many aspects, including DNA transcription, translation and repair, cell construction, intracellular component transportation, and cell synthesis of important molecules. In addition, they also discovered some interactions between proteins with known functions and proteins with unknown functions, laying the foundation for future research on proteins at these locations.
The results of this research, and the major breakthrough in the field of monomer protein structure prediction, indicate that structural biology has entered a new era in which computing plays an important role. Such methods can be further applied to human proteins. Identifying unknown protein complexes in human cells and predicting their structure may provide new treatments for many diseases. In the not-too-distant future, combining the long-term accumulation of experimental data and artificial intelligence methods in academia, scientists will be able to accurately predict the interactions between all proteins in an organism and the three-dimensional structure of all protein complexes. They call this Protein Interactomics. The complete interacting set will clarify many basic problems in biology, and the interface of these protein interactions may be a target to provide a large number of new drugs in the future.
Reference
- Humphreys, I. R., Pei, J., Baek, M., Krishnakumar, A., Anishchenko, I., Ovchinnikov, S., Zhang, J., Ness, T. J., Banjade, S., Bagde, S. R., Stancheva, V. G., Li, X. H., Liu, K., Zheng, Z., Barrero, D. J., Roy, U., Kuper, J., Fernández, I. S., Szakal, B., Branzei, D., … Baker, D. (2021). Computed structures of core eukaryotic protein complexes. Science (New York, N.Y.), eabm4805. Advance online publication.
- BioArt Posts
- Jumper, J., Evans, R., Pritzel, A., Green, T., Figurnov, M., Ronneberger, O., Tunyasuvunakool, K., Bates, R., Žídek, A., Potapenko, A., Bridgland, A., Meyer, C., Kohl, S., Ballard, A. J., Cowie, A., Romera-Paredes, B., Nikolov, S., Jain, R., Adler, J., Back, T., … Hassabis, D. (2021). Highly accurate protein structure prediction with AlphaFold. Nature, 596(7873), 583–589.
- Baek, M., DiMaio, F., Anishchenko, I., Dauparas, J., Ovchinnikov, S., Lee, G. R., Wang, J., Cong, Q., Kinch, L. N., Schaeffer, R. D., Millán, C., Park, H., Adams, C., Glassman, C. R., DeGiovanni, A., Pereira, J. H., Rodrigues, A. V., van Dijk, A. A., Ebrecht, A. C., Opperman, D. J., … Baker, D. (2021). Accurate prediction of protein structures and interactions using a three-track neural network. Science (New York, N.Y.), 373(6557), 871–876.
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.