ProDy - Elastic Network Model
Introduction of Elastic Network Model (ENM) and ProDy package

1. Brief Introduction
The ProDy tool was developed by the Ivet Bahar Laboratory of the University of Pittsburgh, USA, for structure-based protein dynamic analysis. ProDy is a free and open source Python software package. It is designed as a flexible and responsive API suitable for interactive use and the development of computing programs. Users can download it on the ProDy official website and consult the manual. At the same time, after years of development, the ProDy tool has added multiple functions compared to the original version, which can be used for more in-depth study of protein dynamics.
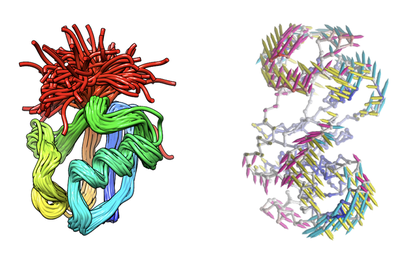
In related publications, the function and application of the tool are described as follows:
“ProDy allows for quantitative characterization of structural variations in heterogeneous datasets of structures experimentally resolved for a given biomolecular system, and for comparison of these variations with the theoretically predicted equilibrium dynamics. Datasets include structural ensembles for a given family or subfamily of proteins, their mutants and sequence homologues, in the presence/absence of their substrates, ligands or inhibitors. Numerous helper functions enable comparative analysis of experimental and theoretical data, and visualization of the principal changes in conformations that are accessible in different functional states.”
In short, the biggest feature of ProDy is to infer the dynamic properties of the protein from theory and experiment. It can provide information about the structural variation of the target system, and make systematic comparisons with the internal dynamics predicted by theoretical models and methods, thereby helping in-depth understanding of the relationship between structure, dynamics and function. The realization of ProDy relies on the proposal of the elastic network model (ENM) theory.
2. Development of Elastic Network Model (ENM)
Molecular Dynamics (MD) simulation is an effective tool to obtain the microscopic motion process of protein at the atomic level. However, since MD simulation is very time-consuming, there are still some difficulties in using MD to simulate the large-scale conformational movement of protein functionality.
In the 1980s, Brooks et al., Go et al. and others developed the Normal Mode Analysis (NMA) method and successfully applied it to the study of protein movement patterns. The NMA method can obtain the conformational motion properties of the system without a long-term dynamic simulation, which greatly reduces the amount of calculation. However, the traditional NMA method needs to use the classical all-atomic force field to optimize the energy of the system. If the energy optimization is not sufficient, the results will be greatly biased, and the calculation of NMA requires a large amount of memory.
In 1996, Tirion et al. greatly simplified the traditional all-atom NMA method, further reducing the amount of calculation. Moreover, the interaction between protein atoms is simplified to resonance potential, replaced by springs, and the strength coefficients of all springs are the same, ignoring the strength of the interaction between different atoms.
On this basis, Bahar et al. and Hinsen et al. further simplified the NMA method and proposed the Elastic Network Model (ENM). ENM simplifies each residue of the protein to a node and uses its Cα instead. The interaction between the residues is simplified to resonance potential, replaced by springs, and the strength coefficients of all springs are the same. ENM completely ignores the fine interaction between residues and simplifies the traditional atomic level NMA method to the residue level, which greatly reduces the amount of calculation and memory consumption. In the model proposed by Bahar et al., the vibration of the residue near the equilibrium position is considered to be an isotropic Gaussian motion. This model is called the Gaussian Network Model (GNM). The GNM model can calculate the motion amplitude of each residue in different motion modes, but it cannot obtain the direction of its motion.
Subsequently, Atilgan et al. extended GNM, considered the directional information of residue movement, developed the isotropic model into an anisotropic model, and established the Anisotropic Network Model (ANM). Because GNM and ANM are simple and fast to calculate, they have been widely used in the study of protein structure-function relationships such as protein allosteric process, binding-induced conformational changes, prediction and identification of protein complex binding sites, which is conducive to in-depth understanding of protein interaction between internal complexes.
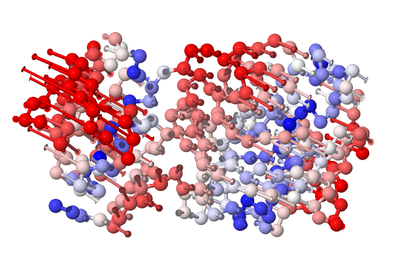
3. Network characterization of ENM
The Elastic Network Model (ENM), as an effective method to obtain the intrinsic dynamics of the protein structure itself, and then reveal its biological functions, has been widely used in the study of protein structure-function relationships. The core idea is to characterize the protein structure with the aid of the network, with the aid of the topological properties of the network model combined with relevant analysis, so as to draw certain conclusions. Regarding why the network model is used, Ivet Bahar Lab gave the following explanation:
√ for large systems' collective motions & long time processes beyond the capability of full atomic simulations
√ to incorporate structural data in the models – at multiple levels of resolution
√ to take advantage of theories developed in other disciplines: polymer physics, graph theory, spectral graph methods, etc.

ProDy applies ENM to practical applications, which helps predict the overall movement of proteins. Although ENM is a coarse-grained description that simplifies each residue to Cα instead, and the residue pairs are connected by elastic springs with uniform strength coefficients, a unique analysis result of the motion spectrum of each protein system can be obtained.
Reference
- Ahmet Bakan, Lidio M. Meireles, Ivet Bahar, ProDy: Protein Dynamics Inferred from Theory and Experiments, Bioinformatics, Volume 27, Issue 11, 1 June 2011, Pages 1575–1577.
- MCCAMMON J A,GELIN B R,KARPLUS M. Dynamics of folded proteins[J]. Nature,1977,267(5612) : 585-590.
- BROOKS B R,KARPLUS M.Harmonic dynamics of proteins: normal modes and fluctuations in bovine pancreatic trypsin inhibitor [J]. Proceedings of the National Academy of Sciences of the United States of America,1983,80(21) : 6571-6575.
- TIRION M M. Large amplitude elastic motions in proteins from a single-parameter,atomic analysis [J]. Physical Review Letters,1996,77(9) : 1905-1908.
- GO N,NOGUTI T,NISHIKAWA T. Dynamics of a small globular protein in terms of low-frequency vibrational modes [J]. Proceedings of the National Academy of Sciences of the United States of America,1983,80(12) : 3696-3700.
- BAHAR I,ATILGAN A R,ERMAN B. Direct evaluation of thermal fluctuations in proteins using a single-parameter harmonic potential[J]. Folding and Design,1997,2( 3) :173-181.
- HINSEN K. Analysis of domain motions by approximate normal mode calculations[J]. Proteins,1998,33(3): 417-429.
- ATILGAN A R,DURELL S R,JERNIGAN R L,et al. Anisotropy of fluctuation dynamics of proteins with an elastic network model[J]. Biophysical Journal,2001,80 (1) :505-515.
- Eyal E,Lum G, Bahar I (2015) The anisotropic Network Model web server at 2015 (ANM 2.0), Bioinformatics 31:1487-9
- Li,H., Chang,YY, Yang,LW and Bahar,I. (2016) iGNM 2.0: The Gaussian Network Model Database for Biomolecular Structural Dynamics. Nucleic Acids Res., 44, D415-D422.
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.