Journal of Chemical Information and Modeling | Comparative Dynamics and Functional Mechanisms of the CYP17A1 Tunnels Regulated by Ligand Binding

Cytochrome CYP17A1 is a bifunctional monooxygenase that binds to cell membranes and plays a key role in the synthesis of many human steroid hormones. The 17α-hydroxylase activity of this enzyme is essential for the production of glucocorticoids. Therefore, CYP17A1 is an important target for the treatment of breast and prostate cancers that are proliferated by estrogen and androgens. In 2012, Emily Scott et al. resolved the first crystal structure of CYP17A1 combined with abiraterone, which provided important structural information for the development of CYP17A1 inhibitory drugs. Although abiraterone was approved for the treatment of metastatic prostate cancer in 2011, patients taking abiraterone usually require other drugs to control high blood pressure, edema and cardiovascular side effects. On the other hand, the active center of CYP17A1 is located inside the protein. In order for the substrate molecule to bind to it, it must pass through the gate on the surface of the protein and the channel connecting the inside and outside. Therefore, the importance of these gates and channels for the specific binding of substrates is self-evident, and there are relatively few studies on them as potential drug targets. It can be seen that the research on how ligand binding affects the dynamics and functional mechanism of CYP17A1 small molecule channels provides new opportunities and challenges for drug development based on its structure.
In the previous work, our research group proposed an integrated genome analysis, co-evolution and biological network method for the modeling of protein dynamics big data, which provides a computational framework for the study of quantitative protein dynamics and allosteric regulation. Recently, our group and Prof. Gennady Verkhivker of Chapman University have expanded this computational strategy to combine dynamic simulation, structural channel modeling, dynamic residue network (DRN) and perturbation response scanning (PRS) methods. The geometry, dynamics, and allosteric control mechanism of the channel have undergone a series of quantitative analysis.
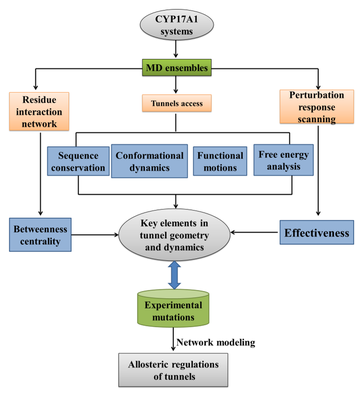
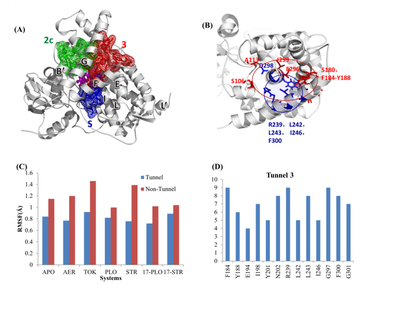
First, the author selected 6 different ligands to bind to CYP17A1 complex system, including 2 drug molecules abiraterone and TOK-001, 2 natural substrates progesterone and pregnenolone (STR/PLO) and both The 17-α hydroxylation products (17-STR, 17-PLO). The dynamic simulations of these six complex systems and the empty system (APO) were carried out, and the channel clustering and geometric property analysis were performed to determine the main channels through which small molecules enter and exit in each system. The clustering results show that the 3 and 2c channels are the main channels shared by the 7 systems, and the amino acid composition of each channel is basically the same.
According to the analysis of dynamic fluctuation and sequence conservation, it is found that the channel residues show stronger rigidity and more significant sequence conservation. In addition, combined with the energy calculation of MM-PBSA, it further shows that the bottleneck residue plays a vital role in the stability, geometric characteristics and ligand binding of the channel.
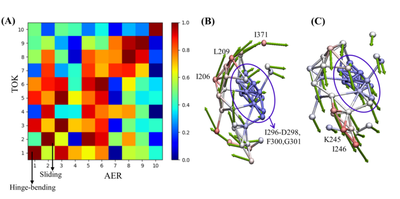
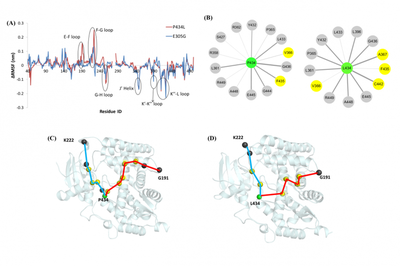
In order to understand the effect of the ligand binding process on the protein channel, in addition to the analysis of the channel bottleneck residues, the author also further explored the movement mode of the channel. The author selects channel 3, which is very important in most systems, as the object, and uses the elastic network model (ANM) method to calculate the cooperative motion mode of this channel in different ligand binding systems. ANM analysis results show that: hinge bending and clip sliding may be the main functional movement modes of the CYP17A1 channel, and there are some common key residues in different systems, which mediate and regulate the functional movement of ligands effect.
In addition, the author also used DRN and PRS to quantitatively analyze protein dynamics. Based on molecular dynamics simulation data, a dynamic residue network was constructed, and it was found that the shortest path parameters of the network can quantitatively describe the conformational changes of the CYP17A1 channel as a whole. The calculation and comparison of the betweenness centrality of the network confirmed that this topology parameter can be used to predict the key regions and residues involved in signal transduction on the channel. In the PRS analysis, these key regions and residues received functional responses in terms of allosteric effects, such as E305 and P434. In fact, experiments have confirmed that mutations at these two sites will result in a significant decrease in the enzyme activity of CYP17A1.
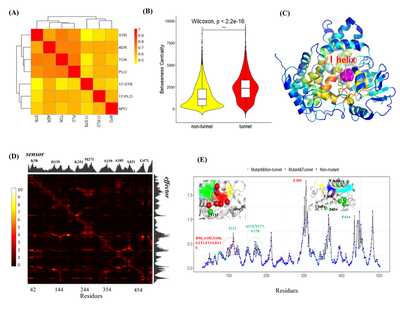
Finally, the mutation system of these two sites (E305G, P434L) was analyzed for network differences, including local changes in the interaction network and analysis of long-distance signal transmission paths. The results show that the mutation site can be used as an allosteric hotspot, and its mutation can cause significant network reorganization, thereby regulating the dynamic properties of the internal channel of CYP17A1 and having a significant impact on the function of CYP17A1. In particular, the non-channel site P434L mutation enhances the long-distance communication with certain surface residues (such as K222, G191), which means that these peripheral residues may in turn control the dynamic behavior of the channel.
To sum up, in this publication, an integrated computational strategy is proposed to be applied to the CYP17A1 complex system combining different ligands, which can systematically study the mechanism of ligand binding and channel tunneling dynamics at the atomic level. The results of this study are consistent with experimental data and provide valuable insights for understanding the structure and function of protein molecular channels. The results and methods also provide a roadmap for the prediction of allosteric mechanisms and the discovery of CYP enzyme drugs.
Reference
- DeVore, N. M.; Scott, E. E. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012, 482, 116-119
- Liang, Z.; Verkhivker, G. M.; Hu, G. Integration of network models and evolutionary analysis into high-throughput modeling of protein dynamics and allosteric regulation: theory, tools and applications. Briefings Bioinf. 2020, 21, 815– 835
- Xiao, F.; Song, X.; Tian, P.; Gan, M.; Verkhivker, G. M.; Hu, G. Comparative Dynamics and Functional Mechanisms of the CYP17A1 Tunnels Regulated by Ligand Binding. J. Chem. Inf. Model. 2020, 60, 3632-3647
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.