Nature Communications | CopulaNet - Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction

In the past few years, significant progress has been made in the field of protein structure prediction. Among them, the prediction of contact/distance between residues has played an important role. Most recent protein structure prediction methods, such as AlphaFold and trRosetta, use roughly the same three-step process: i) estimate the contact/distance between residues; ii) construct the potential energy function based on the estimated contact/distance; iii) establish a three-level structure with the smallest potential energy. These methods of estimating the contact/distance between residues usually require constructing a multiple sequence alignment (MSA) for the target protein, and then performing residue co-evolution analysis on the resulting MSA. The basic principle is that two residues that are close in space always tend to co-evolve, so in turn, the co-evolution of residues can be used to accurately estimate the contact/distance between residues.
In order to derive the direct coupling of residues, various direct coupling analysis (DCA) methods using precision matrix or Potts model have been widely used. Although the DCA technique has been proven effective in estimating the contact/distance between residues, it still has some disadvantages. A significant shortcoming is that a large amount of information is lost after converting MSA to manually extracting features, that is, the covariance matrix. In fact, the premise of the DCA technique is that the direct coupling between two residues can be modeled by using paired statistics (such as covariance). However, this premise is not always true. The author proposes two artificial proteins P1 and P2 as counterexamples. In protein P1, the two residues R1 and R2 are close, while in protein P2, they are far away from each other. Although there are big differences in the MSA structure of P1 and P2, the covariance matrices calculated from these MSAs are exactly the same, resulting in the DCA technique giving the same distance estimation for proteins P1 and P2. Therefore, the author believes that the method of extracting direct coupling between residues from MSA is more necessary and effective.
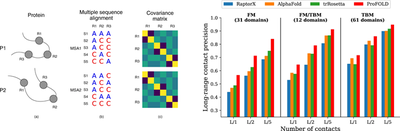
In this article, the author reports an end-to-end deep neural network framework called CopulaNet for estimating the distance between residues. CopulaNet learns the conditional joint residue distribution directly from MSA, rather than learning from manual features such as covariance matrices. CopulaNet is composed of three key parts, namely MSA encoder, co-evolution aggregator and distance estimator. The MSA encoder processes the K homologous proteins of the target protein separately to obtain K alignments, and uses a one-dimensional convolution residual network with multiple convolution layers to obtain the embedding feature for each residue mutation containing the information of neighboring residues. For any two residues, the aggregator first calculates the outer product derived from the embedding feature of each homologous protein, and then uses the average pool to aggregate the outer product obtained from all homologous proteins, and obtains the commonality between the two residues to get information about co-evolution between two residues. Based on the obtained co-evolution, the author finally uses a two-dimensional residual network to estimate the distance of residue pairs. They divided the distance range between residues into 37 intervals and converted the distance estimation problem into a classification problem.
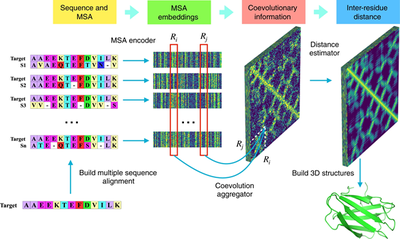
Using CopulaNet as the core module, the author developed a method for predicting protein structure called ProFOLD. ProFOLD converts the estimated distance into a potential function and obtains a three-level structure with the smallest potential energy. The author first uses the protein T0992-D1 as an example to illustrate the concept of ProFOLD, and then uses it to predict the structure of the CASP13 target protein and compares it with other latest prediction methods, showing the superiority of ProFOLD and trying to analyze the contribution of the key elements of CopulaNet.
All source codes and models of ProFOLD are publicly available through https://github.com/fusong-ju/ProFOLD.
Reference
- Ju, F., Zhu, J., Shao, B. et al. CopulaNet: Learning residue co-evolution directly from multiple sequence alignment for protein structure prediction. Nat Commun 12, 2535 (2021).
Yu Zhu
MSc Drug Design student
My research interests include bioinformatics, computational biology and systems biology, especially in protein related biological problems.